Usage¶
Preparation¶
STRique works on raw nanopore read data in either single or bulk fast5 format. Batches of single reads in tar archives work as well. Before starting the repeat detection the raw data folder must be indexed to enable extraction of single reads. The index file contains relative paths to the reads and must be saved in the indexed directory.
python3 scripts/STRique.py index [OPTIONS] input
positional arguments:
input Input batch or directory of batches
optional arguments:
--recursive Recursively scan input
--out_prefix OUT_PREFIX Prefix for file paths in output
--tmp_prefix TMP_PREFIX Prefix for temporary data
The command to recursively index the raw data archive could look similar to the following. Indexing is only required once after the sequencing is completed.
python3 ~/src/STRique/scripts/STRique.py index \
--recursive ~/my_data > ~/my_data/reads.fofn
The index file reads.fofn contains relative paths to the raw files and must therefore be saved in the right location. You can configure an --out_prefix to be prepended to each entry in the index. This is useful to index sub-directories while storing indices at a central location e.g.:
python3 ~/src/STRique/scripts/STRique.py index \
--recursive --out_prefix my_sample ~/my_data/my_sample > ~/my_data/my_sample.fofn
Configuration¶
Targeted repeats are configured in a tab-separated (.tsv) file with columns
chr begin end name repeat prefix suffix
The file must have the header line and can contain as many repeats as present/targeted by enrichment. For the hexanucleotide repeat at the c9orf72 locus the (truncated) config for hg19 alignments looks like this (A complete example file is in the config folder of the STRique repository):
chr9 27573527 27573544 c9orf72 GGCCCC ...GCCCCGACCACGCCCC TAGCGCGCGACTCCTG...
STRique will only consider aligned reads where the mapping including soft-clipping at least partially covers one of the configured targets. The longer the prefix/ suffix sequences, the more reliable the signal alignment at the cost of a longer runtime. A good estimate is 150 Bp for prefix and suffix. Repeat, prefix and suffix sequence are always on template strand.
Repeat Counting¶
The repeat detection requires an indexed raw data archive and the alignment of the reads:
python3 scripts/STRique.py count [OPTIONS] f5Index model repeat
positional arguments:
f5Index Fast5 index
model pore model
repeat repeat region config file
optional arguments:
--out OUT output file name, if not given print to stdout
--algn ALGN alignment in sam format, if not given read from stdin
--mod_model MOD_MODEL Base modification pore model
--config CONFIG Config file with HMM transition probabilities
--t T Number of processes to use in parallel
--log_level Log level {error,warning,info,debug}
The command to detect repeat lengths could look similar to:
cat ~/my_data.hg19.sam | python3 ~/src/STRique/scripts/STRique.py count \
~/my_data/reads.fofn ~/src/STRique/models/r9_4_450bps.model \
~/src/STRique/configs/repeat_config.tsv \
> ~/my_data.hg19.strique.tsv
For Docker users the container needs to mount the host file system to access the raw data. To process the same dataset as above run:
docker run -it --mount type=bind,source=${HOME},target=/host \
giesselmann/strique:v0.3.0
cat /host/my_data.hg19.sam | python3 scripts/STRique.py count \
/host/my_data/reads.fofn models/r9_4_450bps.model \
configs/repeat_config.tsv > /host/my_data.hg19.strique.tsv
Please note that changes made to the Docker filesystem are not persistent and will be lost after exiting the container. Make sure to write the output to a file on the host.
Output¶
The output of STRique is a tsv file with repeat counts per read covering at least one region specified in the repeat config file. Reads are reported as soon as they overlap one of the regions, even if the repeat detection was not successful or not reliable. It is therefore suggested to filter the raw output. The columns of the output stream are as follows:
- ID: The read identifier from MinKNOW
- target: The repeat expansion target specified in the config file
- strand: + or -
- count: Repeat count as number of occurrences of the configured repeat sequence
- score_prefix and score_suffix: Signal alignment scores for post processing
- log_p: HMM log-likelihood over the repeat, generally larger for longer repeats
- offset, ticks: Offset and length of the repeat in the raw signal fast5 file, for debugging and visualization
- mod: Base modification string if --mod_model was provided e.g. '0001000' for a mostly unmethylated repeat
In a first step reads with zero repeat count can be discarded, this happens for instance, if prefix and suffix sequence are found in the wrong order in the raw signal. Secondly it is reasonable to further filter for the signal alignment scores score_prefix and score_suffix. These are indicators for the quality of the prefix and suffix mappings and have strong impact on the subsequent repeat counting. For the c9orf72 and FMR1 samples tested in our publication, a threshold of 4.0 was chosen. This value might change for other targets and configurations and is in general a tradeoff between number of evaluated repeats and their quality.
The base modification string contains one character per repeat instance with zeros for the base model and ones for the modification model to be more likely. Since the HMM for repeat base modifications is slightly different, the length of the string is not always exactly equal to the reported repeat count. It is recommended to further compute a per read mean repeat methylation.
Masking¶
STRique detects the repeat expansion in the raw nanopore signal. It is possible to modify .fast5 files and mask the repeat, in order to feed the data into conventional downstream pipelines. The command line of the masker-script is as follows:
python3 scripts/fast5Masker.py [OPTIONS] index counts output
Mask region in raw nanopore fast5 file.
positional arguments:
index Path to input fast5 index
counts Path to STRique count output file
output Path to output .fast5 directory with masked reads
optional arguments:
-h, --help show this help message and exit
--format {single,bulk} Output fast5 format
For example:
python3 ~/src/STRique/scripts/fast5Masker.py ~/my_data/reads.fofn ~/my_data.hg19.strique.tsv ~/my_data_masked/
Output files can either be written as single- or bulk-fast5, depending on your needs.
Plot¶
STRique comes with a basic visualization of the repetitive signal. After raw indexing and counting you can plot the raw nanopore traces of the repeat with the following command:
python3 scripts/STRique.py plot [OPTIONS] f5Index
positional arguments:
f5Index Fast5 index
optional arguments:
-h, --help show this help message and exit
--counts COUNTS Repeat count output from STRique, if not given read from stdin
--output OUTPUT Output directory for plots, use instead of interactive GUI
--format Output format when writing to files {pdf,svg,png}, default=png
--width WIDTH Plot width, default=16
--height HEIGHT Plot height, default=9
--dpi DPI Resolution of plot, default=80
--extension EXTENSION Extension as fraction of repeat signal around STR region to plot
--zoom ZOOM Region around prefix and suffix to plot
--log_level Log level {error,warning,info,debug}
For example:
cat ~/my_data.hg19.strique.tsv | python3 ~/src/STRique/scripts/STRique.py plot ~/my_data/reads.fofn
You can either read the STRique counts from stdin or specify a file with the --counts flag. The plots get displayed in a matplotlib GUI. Alternatively you can specify --output to write each plot to a separate file. The filename is target_count_ID.format. The output for the provided example data looks like this:
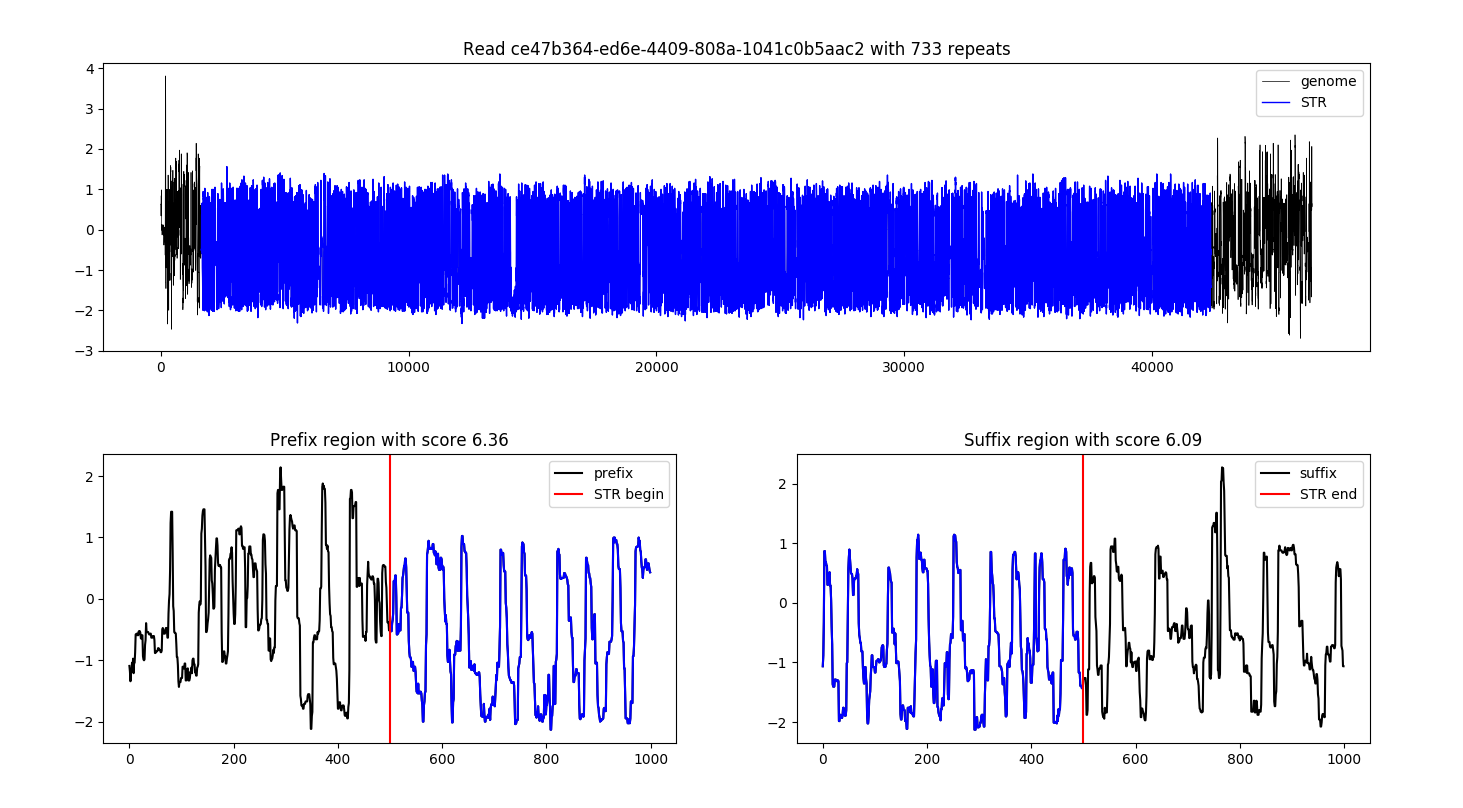
Docker
The plot commands tries to open an interactive GUI per default. If you use STRique from within a Docker container please specify the --output argument to write the plots to disk. From within the container it is non-trivial to forward graphical output to the host.